长沙市中医医院 | 脓毒症多器官炎症的 “克星”?研究揭示 FGF15 调控 H3K18 乳酸化与 Irf7 表达的关键机制
- 2026-06-08 03:28:27

导读
为解决脓毒症缺乏特异性免疫靶向治疗的问题,本研究以 M1 巨噬细胞极化驱动脓毒症炎症为核心切入点,通过体内 CLP 脓毒症小鼠模型与体外 LPS 刺激巨噬细胞实验,探究 rFGF15 的抗炎作用及机制;先验证 rFGF15 对巨噬细胞糖酵解、组蛋白乳酸化及 M1 极化的影响,再通过通路干预、基因沉默 / 过表达等明确 NF2-Hippo 通路的介导作用,最终通过巨噬细胞移植实验验证体内疗效,结论为 rFGF15 可通过 FGFR4 激活 NF2-Hippo 通路,抑制糖酵解与 H3K18 乳酸化,减少 Irf7 表达,从而抑制 M1 巨噬细胞极化及多器官炎症,为脓毒症提供潜在治疗方案。
研究亮点
本研究首次揭示 FGF15 调控脓毒症 M1 巨噬细胞极化的表观遗传机制,将 FGFR4-NF2-Hippo 通路、糖酵解代谢、H3K18 乳酸化与 Irf7 表达串联成完整调控链;通过体内外实验结合巨噬细胞移植,验证 rFGF15 可改善脓毒症小鼠多器官炎症与生存率;明确 H3K18 乳酸化对 Irf7 启动子的靶向调控作用,为脓毒症的代谢 - 表观遗传联合干预提供新思路,凸显 FGF15 作为创新治疗靶点的潜力。
全文归纳总结


研究方法
动物实验:采用 C57BL/6 小鼠构建 CLP 脓毒症模型,分为假手术组、CLP 组、CLP+rFGF15 组,通过尾静脉注射 rFGF15 干预,检测多器官炎症与生存情况;采用脂质体氯膦酸盐耗竭巨噬细胞后,移植经不同处理的骨髓来源巨噬细胞(BMDMs),评估移植效果。
细胞实验:分离小鼠 BMDMs 及培养 RAW264.7 巨噬细胞,经 LPS 刺激构建脓毒症细胞模型,给予 rFGF15 及 FGFR4、NF2 沉默质粒、Irf7 过表达质粒等干预;
检测技术:运用 Western blot 检测蛋白表达、HPLC-MS/MS 分析糖酵解代谢物、流式细胞术检测巨噬细胞极化与免疫细胞比例、ELISA 测定细胞因子水平、ChIP-qPCR 与 CUT&Tag 验证组蛋白修饰与基因结合、Co-IP 验证蛋白相互作用、qRT-PCR 检测基因表达、组织病理学分析器官损伤。
研究结果
rFGF15 可恢复 CLP 小鼠巨噬细胞中降低的 p-FGFR4/FGFR4 比值,抑制 LPS 诱导的巨噬细胞糖酵解,降低糖酵解代谢物水平、葡萄糖摄取及乳酸产生,下调糖酵解关键酶与 MCT1 表达;
rFGF15 减少脓毒症巨噬细胞中总组蛋白乳酸化及 H3K18la 水平,该效应可被 FGFR4 沉默逆转;
rFGF15 抑制 M1 巨噬细胞极化,降低促炎因子(TNF-α、IL-1β 等)水平,升高抗炎因子(IL-10、TGF-β 等),改善脓毒症小鼠多器官炎症浸润与功能损伤;
机制上,rFGF15 通过 FGFR4 与 NF2 结合并磷酸化 NF2,激活 Hippo 通路,NF2 沉默或突变可阻断 rFGF15 的上述作用;
H3K18la 富集于 Irf7 启动子促进其表达,rFGF15 可抑制该富集效应,Irf7 过表达会逆转 rFGF15 对 M1 极化的抑制;
巨噬细胞移植实验显示,rFGF15 预处理的 BMDMs 可改善脓毒症小鼠生存率,降低细菌负荷与多器官损伤。
研究结论
本研究证实,rFGF15 可通过 FGFR4 激活 NF2-Hippo 信号通路,抑制脓毒症巨噬细胞的糖酵解代谢,减少乳酸产生与 H3K18 组蛋白乳酸化,进而抑制 Irf7 基因表达,最终阻断 M1 巨噬细胞极化与过度炎症反应,减轻脓毒症小鼠多器官功能损伤并提高生存率。该研究明确了 FGF15 在脓毒症中的抗炎作用及分子机制,将代谢重编程与表观遗传调控相结合,为脓毒症的治疗提供了新的靶点与理论依据,表明 FGF15 有望成为脓毒症的创新治疗药物。
局限性:未探究非组蛋白乳酸化在脓毒症中的作用,未明确 H3K18la 与 Irf7 启动子的具体结合序列,未涉及 NF2-Hippo 通路对补体系统等其他通路的影响。展望:后续将通过生物信息学分析明确 H3K18la 与 Irf7 的结合位点,探究非组蛋白乳酸化的作用,以及 rFGF15-Hippo 通路对补体系统的调控,为 FGF15 临床转化提供更全面的依据。
结果图解


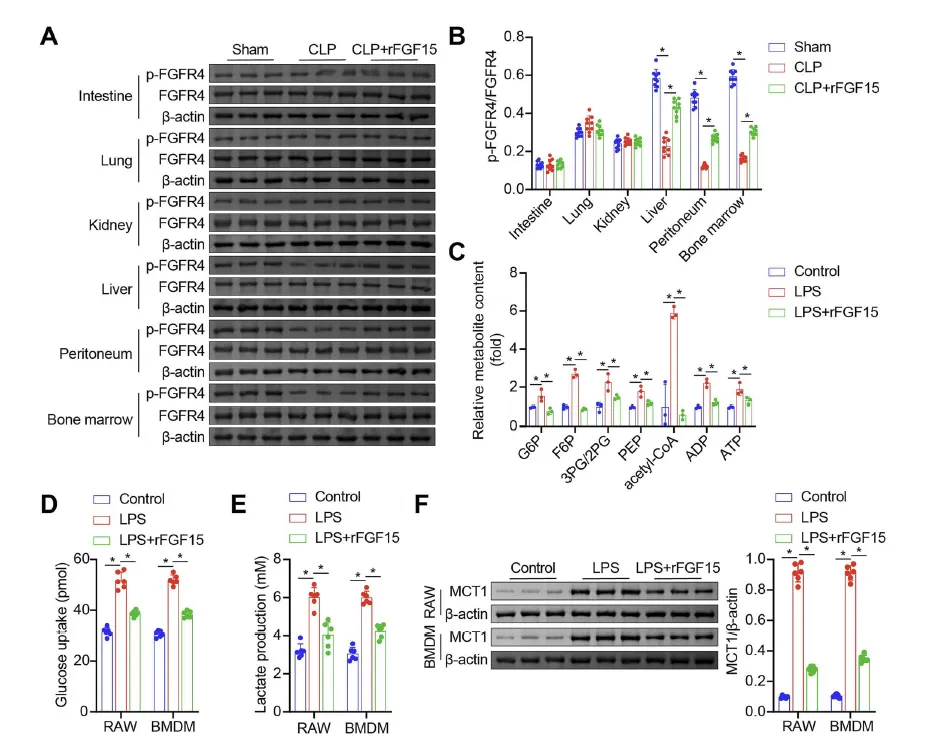
Figure 1:FGF15/FGFR4 信号通路抑制脓毒症巨噬细胞的糖酵解过程
该图通过体内外实验结合多种检测技术,明确了 FGF15/FGFR4 信号对脓毒症巨噬细胞糖酵解的调控作用。体内实验中,Western blot 结果显示 CLP 脓毒症小鼠肝脏、腹膜和骨髓来源的巨噬细胞中磷酸化 FGFR4(p-FGFR4)与 FGFR4 的比值显著下调,而尾静脉注射 rFGF15 可恢复该比值,表明脓毒症状态下 FGF15/FGFR4 信号被抑制,外源性 rFGF15 能激活该信号。体外实验中,HPLC-MS/MS 分析发现 LPS 刺激的小鼠 BMDMs 中糖酵解代谢物(G6P、F6P、3PG/2PG 等)水平显著升高,而 rFGF15 处理可使这些代谢物水平恢复正常;同时,LPS 刺激会增强 BMDMs 和 RAW264.7 巨噬细胞的葡萄糖摄取能力、乳酸产生量,上调糖酵解关键酶(如 HK 系列、PFK 系列、LDH 系列)和乳酸跨膜转运限速蛋白 MCT1 的表达,这些效应均能被 rFGF15 逆转,证实 FGF15/FGFR4 信号可通过抑制糖酵解相关代谢物、关键酶及转运蛋白,阻断脓毒症巨噬细胞的糖酵解异常激活。
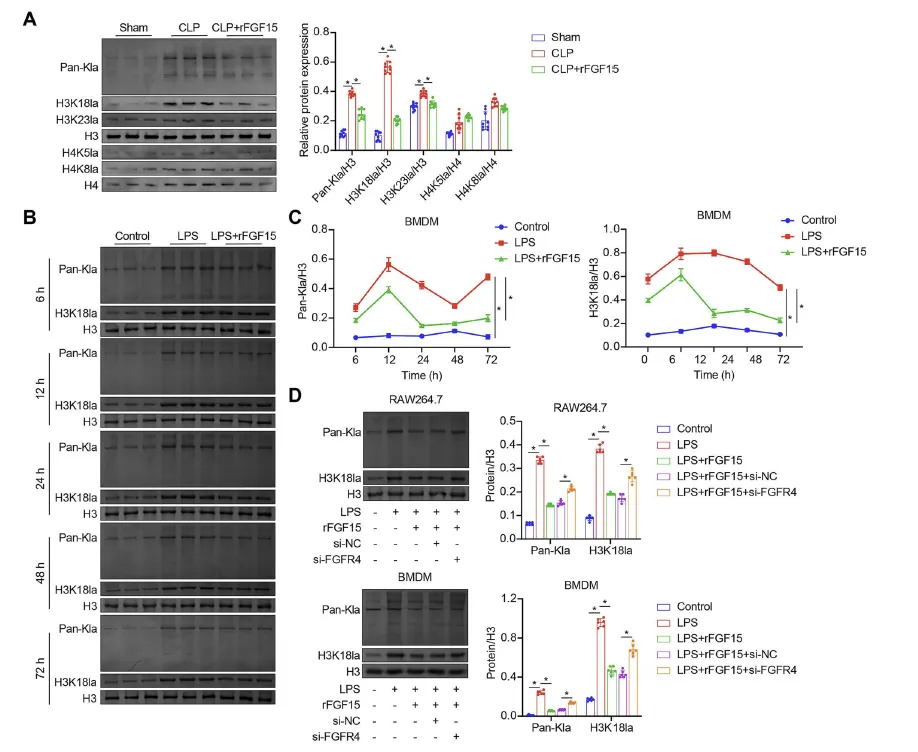
Figure 2:FGF15/FGFR4 信号通路下调脓毒症巨噬细胞的组蛋白乳酸化水平
该图聚焦组蛋白乳酸化这一表观遗传修饰,探究了 FGF15/FGFR4 信号的调控作用。体内实验中,Western blot 检测显示 CLP 脓毒症小鼠 BMDMs 中总组蛋白乳酸化(Pan-Kla)及特异性位点 H3K18la、H3K23la 的水平显著升高,rFGF15 处理能有效降低这些乳酸化蛋白的表达,且对 H3K18la 的调控效果更为显著。体外实验中,时间梯度检测发现 LPS 刺激 BMDMs 后,Pan-Kla 和 H3K18la 水平随时间延长逐渐升高,而 rFGF15 处理可明显抑制这一趋势;进一步通过 FGFR4 沉默实验证实,rFGF15 对组蛋白乳酸化的下调作用依赖 FGFR4,当 FGFR4 被沉默后,rFGF15 无法降低 LPS 诱导的 Pan-Kla 和 H3K18la 升高,表明 FGF15/FGFR4 信号通路可特异性调控脓毒症巨噬细胞的组蛋白乳酸化,尤其针对 H3K18la 位点。
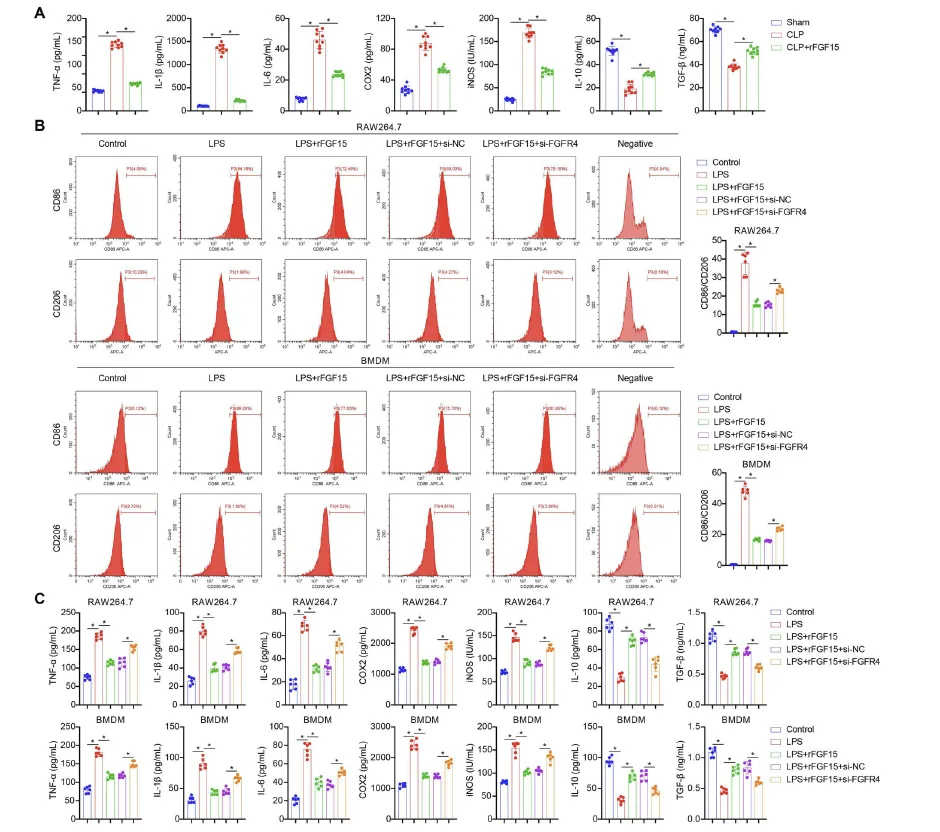
Figure 3:FGF15/FGFR4 信号通路抑制脓毒症巨噬细胞的 M1 极化及炎症反应
该图通过体内外炎症指标检测和细胞表型分析,阐明了 FGF15/FGFR4 信号对巨噬细胞极化和炎症的调控效应。体内实验中,ELISA 结果显示 CLP 脓毒症小鼠血清中促炎介质(TNF-α、IL-1β、IL-6 等)水平升高,抗炎介质(IL-10、TGF-β)水平降低,而 rFGF15 administration 能逆转这一炎症失衡状态。体外实验中,流式细胞术检测发现 LPS 刺激会使 BMDMs 和 RAW264.7 巨噬细胞中 M1 型标志物 CD86 阳性细胞比例升高,M2 型标志物 CD206 阳性细胞比例降低,导致 M1/M2 比值上升,同时细胞培养体系中促炎介质表达上调、抗炎介质表达下调;而 rFGF15 处理可恢复 M1/M2 平衡,减轻细胞炎症反应,且这一保护作用可被 FGFR4 沉默所阻断,说明 FGF15/FGFR4 信号通路能通过抑制巨噬细胞向 M1 型极化,减少促炎因子释放,从而缓解脓毒症相关炎症。
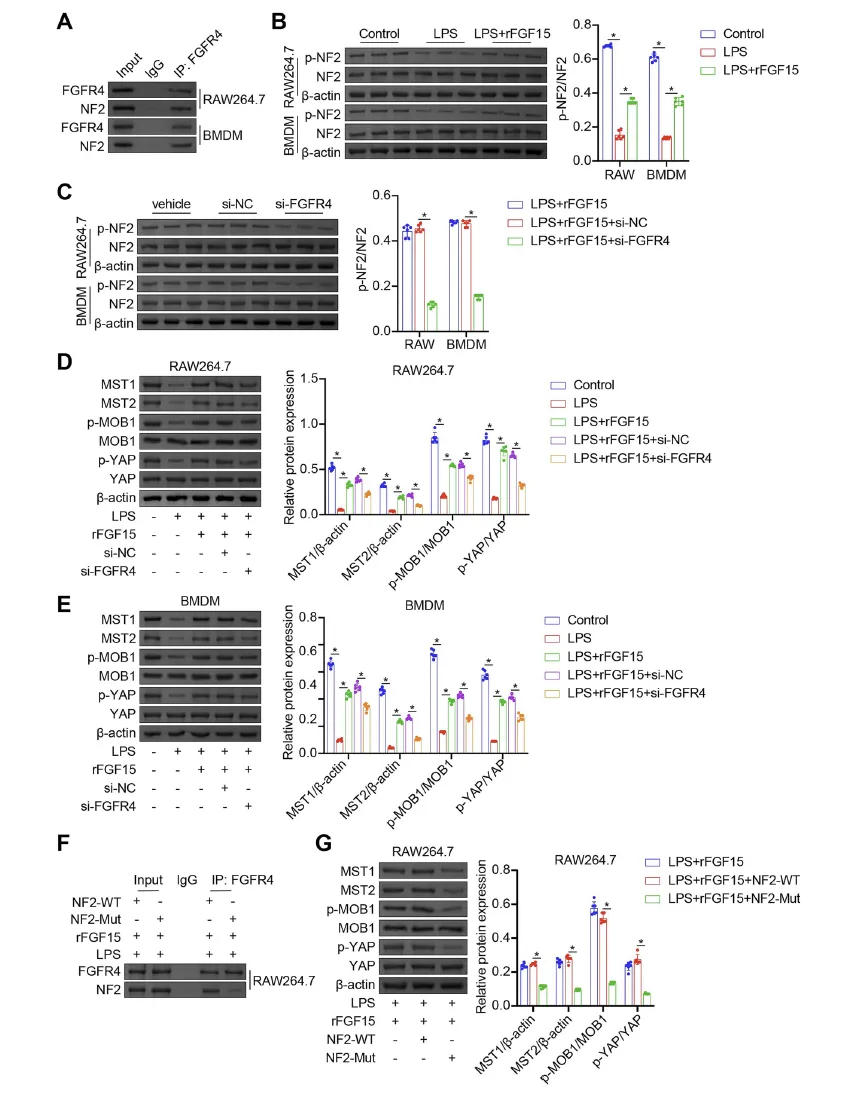
Figure 4:FGF15/FGFR4 信号通路激活脓毒症巨噬细胞的 NF2-Hippo 通路
该图通过蛋白相互作用检测和通路关键分子表达分析,揭示了 FGF15/FGFR4 信号与 NF2-Hippo 通路的调控关系。Co-IP 实验证实,小鼠 BMDMs 和 RAW264.7 巨噬细胞中 FGFR4 与 NF2 存在直接相互作用;Western blot 结果显示,LPS 刺激会降低巨噬细胞中磷酸化 NF2(p-NF2)、MST1/2、磷酸化 MOB1(p-MOB1)和磷酸化 YAP(p-YAP)的水平,表明 NF2-Hippo 通路被抑制,而 rFGF15 处理可恢复这些蛋白的表达,且 FGFR4 沉默会消除 rFGF15 的激活效应。此外,IL-4/IL-13 诱导 M2 极化实验显示,rFGF15 可特异性升高 NF2/MOB1 磷酸化水平和 MST1/2 表达,而对 YAP 磷酸化无明显影响; phospho-null(Y207F)NF2 突变体实验进一步证实,NF2 磷酸化是 FGFR4 与 NF2 结合及 Hippo 通路激活的必要条件,说明 FGF15 通过结合 FGFR4,招募并磷酸化 NF2,进而激活 NF2-Hippo 信号通路。
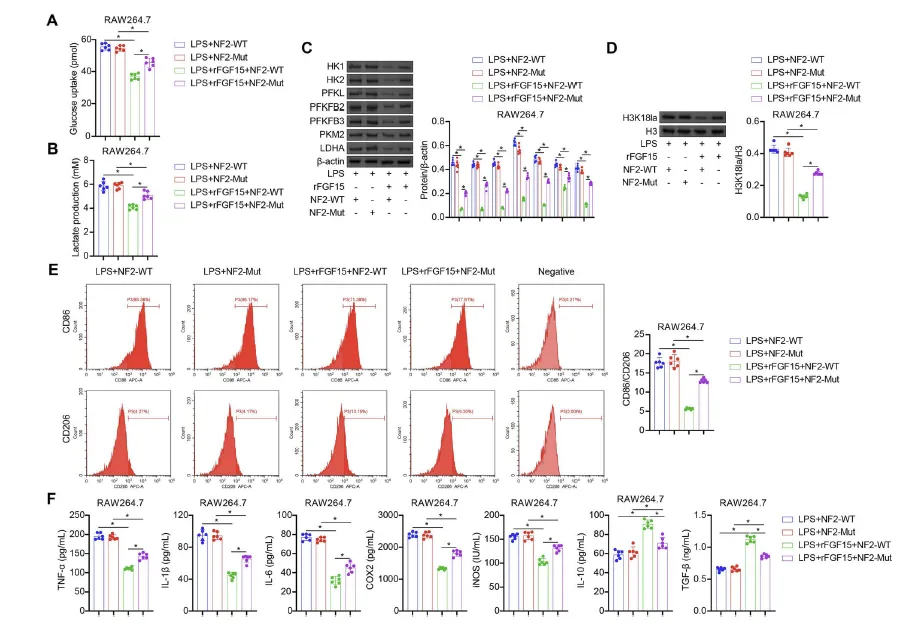
Figure 5:FGF15/FGFR4 信号通路通过 NF2-Hippo 通路调控脓毒症巨噬细胞的糖酵解、组蛋白乳酸化及 M1 极化
该图通过 NF2 野生型(NF2-WT)和突变体(NF2-Mut)转染实验,验证了 NF2-Hippo 通路在 FGF15 调控效应中的介导作用。结果显示,在 RAW264.7 巨噬细胞中,rFGF15 能抑制 LPS 诱导的葡萄糖摄取增加、乳酸产生增多、糖酵解关键酶(HK1、PFKL 等)表达上调及 H3K18la 水平升高,但转染 NF2-Mut 后,rFGF15 的这些抑制效应被完全阻断。同时,流式细胞术和 ELISA 检测发现,rFGF15 对 LPS 诱导的 M1/M2 比值升高及促炎介质释放增加的抑制作用,也因 NF2-Mut 转染而消失;类似地,NF2 沉默(si-NF2)实验在 BMDMs 和 RAW264.7 巨噬细胞中得到一致结果,表明 FGF15/FGFR4 信号通路对糖酵解、组蛋白乳酸化、M1 极化及炎症反应的调控,均依赖于 NF2-Hippo 通路的激活,NF2 是该调控 cascade 中的关键介导分子。
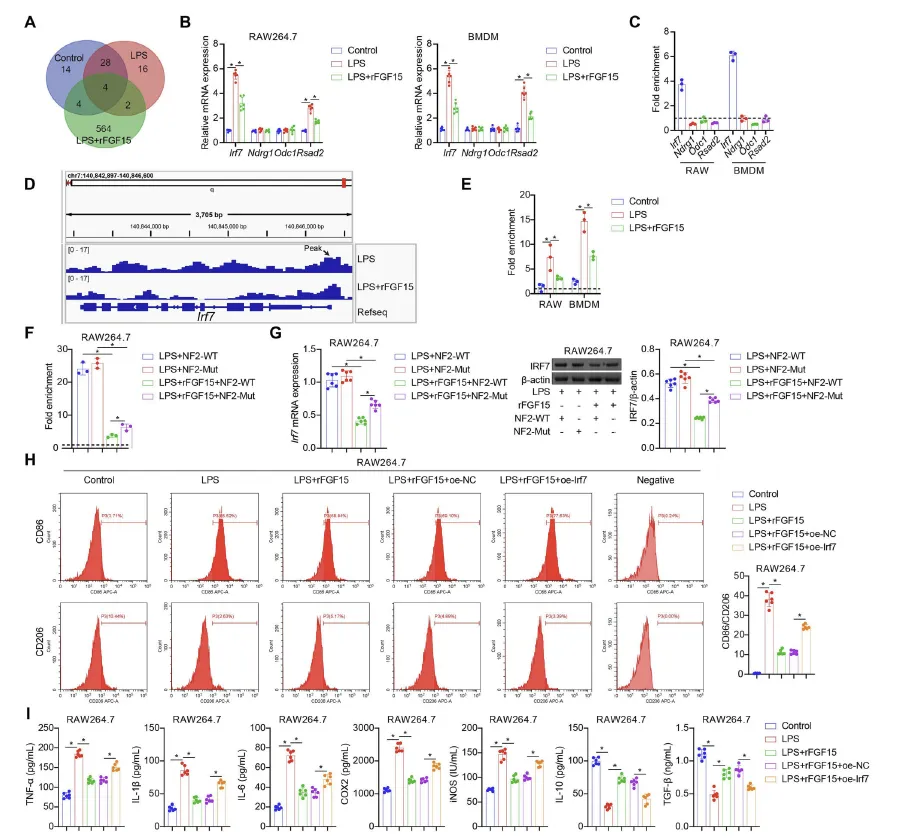
Figure 6:FGF15/FGFR4 信号通路通过 NF2 依赖的 H3K18la 下调抑制 Irf7 表达,进而抑制脓毒症巨噬细胞 M1 极化
该图通过基因表达分析、表观遗传结合验证及功能回复实验,揭示了 FGF15 调控巨噬细胞极化的下游分子机制。RNA-seq 筛选出 LPS 刺激和 rFGF15 处理后差异表达的基因 Irf7 和 Rsad2,qRT-PCR 验证显示 LPS 诱导两者 mRNA 表达升高,rFGF15 可抑制该效应。ChIP-qPCR 和 CUT&Tag 实验证实,LPS 刺激会增强 H3K18la 在 Irf7 启动子区域的富集,而 rFGF15 处理可减少这种富集,且 Rsad2 启动子区域无明显 H3K18la 富集,表明 H3K18la 可特异性调控 Irf7 表达。进一步实验显示,NF2 沉默或 NF2-Mut 转染会减弱 rFGF15 对 H3K18la 富集和 Irf7 表达的抑制作用;而 Irf7 过表达(oe-Irf7)可部分逆转 rFGF15 对 LPS 诱导的 M1 极化和炎症反应的抑制效应,说明 FGF15/FGFR4 信号通路通过 NF2-Hippo 通路抑制 H3K18la 在 Irf7 启动子的富集,降低 Irf7 表达,进而抑制巨噬细胞 M1 极化。
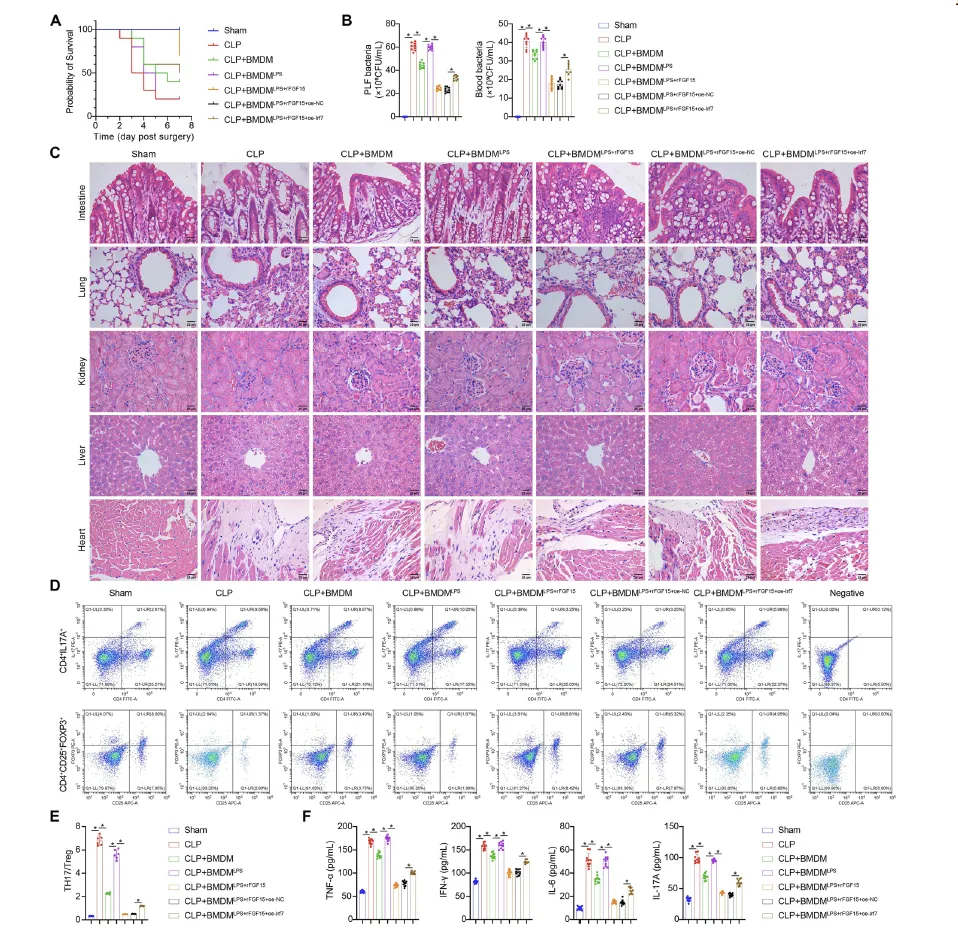
Figure 7:FGF15 通过抑制 Irf7 介导的 BMDM 激活,减轻脓毒症小鼠的多器官炎症并改善生存
该图通过巨噬细胞耗竭 - 移植实验,在体内验证了 FGF15-Irf7-BMDM 调控轴的功能意义。生存分析显示,巨噬细胞耗竭的 CLP 小鼠生存率降低,而移植 rFGF15 预处理的 LPS 刺激 BMDMs 后,小鼠生存率显著提高,且 Irf7 过表达会削弱这一保护作用。细菌负荷检测显示,移植 rFGF15 预处理的 BMDMs 可降低小鼠外周血和腹腔灌洗液中的细菌数量,而 Irf7 过表达会逆转该效应。组织病理学分析显示,CLP 小鼠心、肝、肠、肺、肾等器官存在明显免疫细胞浸润,而移植 rFGF15 预处理的 BMDMs 可减少浸润,Irf7 过表达则会加重器官炎症;同时,rFGF15 预处理的 BMDMs 移植还能改善脓毒症小鼠的肺水肿、肝肾功能损伤、肠道屏障破坏和心脏功能异常,这些保护作用均因 Irf7 过表达而减弱。此外,流式细胞术和 ELISA 检测显示,rFGF15 预处理的 BMDMs 移植可降低小鼠外周血中 Th17/Treg 比值和促炎因子(TNF-α、IFN-γ 等)水平,表明 FGF15 通过抑制 Irf7 介导的 BMDM 异常激活,减轻脓毒症小鼠的多器官炎症损伤,改善整体预后。
#脓毒症#FGF15#巨噬细胞极化#NF2Hippo通路#H3K18乳酸化#Irf73糖酵解#炎症调控

国家杰青一对一答疑视频
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划

