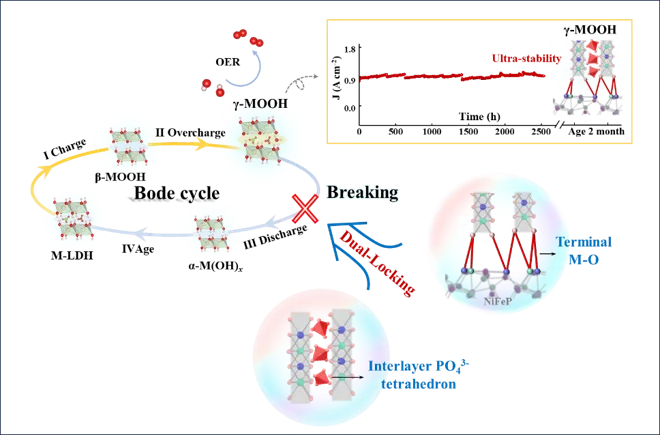
湖南大学黄维清、黄桂芳、万惠(长沙学院)团队ACB文章:打破Bode循环:通过双锁策略稳定亚稳态γ-NiFeOOH用于安培级水氧化
第一作者:冼铭华(湖南大学)
通讯作者:黄桂芳 教授(湖南大学)、万惠 讲师 (长沙学院)、黄维清 教授(湖南大学)
论文DOI: 10.1016/j.apcatb.2026.127185
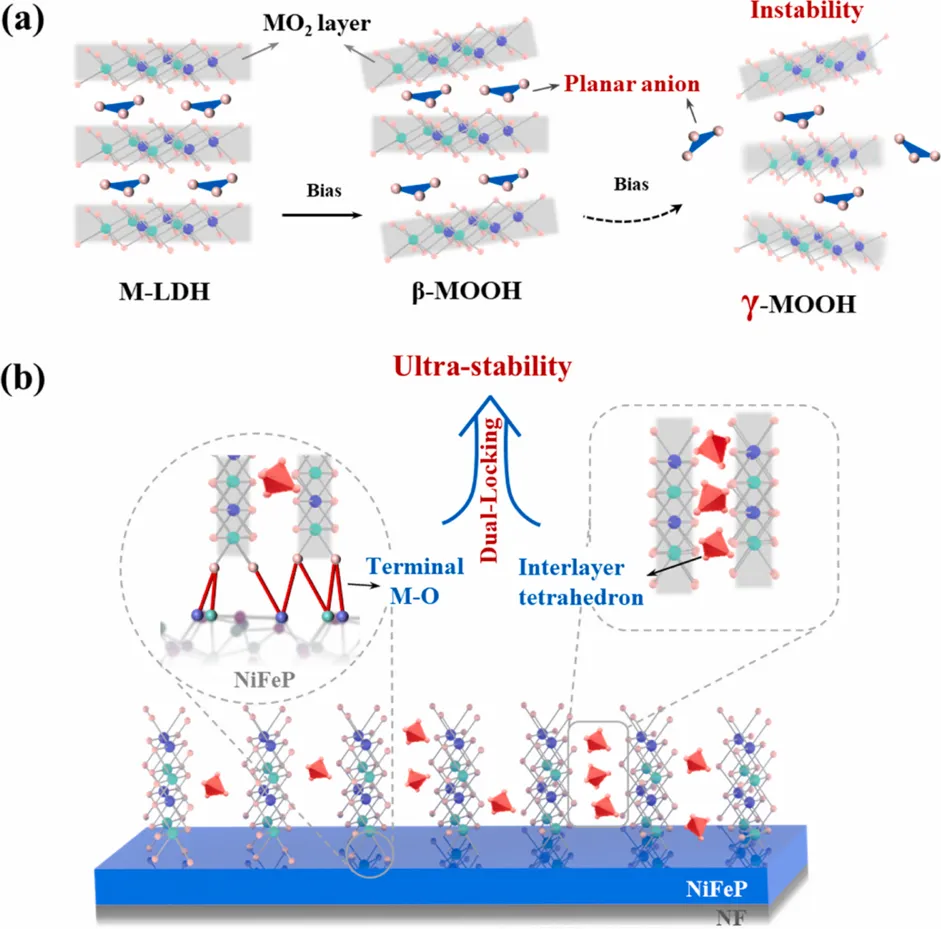
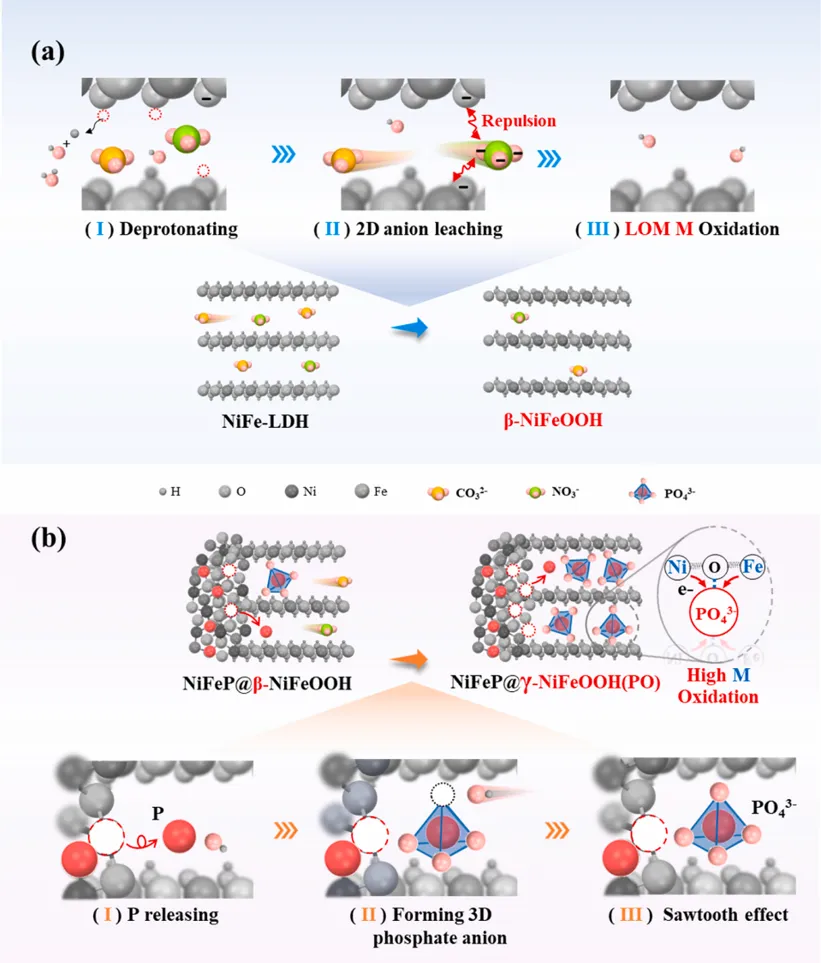
近日,湖南大学黄维清、万惠(长沙学院)、黄桂芳教授团队在Applied Catalysis B: Environment and Energy上发表了题为“Breaking the Bode cycle: Stabilizing metastable γ-NiFeOOH via a dual-locking strategy for ampere-level water oxidation”的研究论文(DOI: 10.1016/j.apcatb.2026.127185)。该工作聚焦碱性水氧化反应(OER)中一个非常关键但长期棘手的问题:高活性γ-NiFeOOH虽然被认为是NiFe基催化剂的重要活性相,但由于其高的氧化态和扩大的层间结构,容易按照经典Bode循环发生结构回退,导致活性衰减和长期稳定性不足。针对这一问题,作者提出了一种“interlayer reinforcement and substrate anchoring”的双重锁定策略。以NiFeP@NiFe-LDH为前驱体,在电化学活化过程中,外层NiFe-LDH重构为NiFeOOH,同时NiFeP内核释放出的P物种迁移进入NiFeOOH层间,并转化为高电荷密度的四面体PO43-基团。这些磷酸根单元像“刚性支柱”一样撑开并咬合相邻Ni(Fe)O2层板,而重构后的层板又通过界面M-O键锚定在导电NiFeP基底上,从而共同稳定亚稳态γ-NiFeOOH。最终得到的NiFeP@γ-NiFeOOH(PO)在100 mA cm-2下仅需236 mV过电位,并可在约900 mA cm-2 (30 °C) 条件下稳定运行超过2500 h以及在长期电解和两个月开路老化后仍保持γ相结构;在约1000 mA cm-2 (60 °C)条件下稳定运行超过350 h;在约1500 mA cm-2 (30 °C)条件下稳定运行超过250 h;同时,在经历10000圈CV和210次循环启停测试后,活性不衰减,为稳定亚稳态高活性催化相提供了新思路。氧析出反应(OER)是电解水制氢的关键半反应,但在工业级电流密度下实现高效且稳定的水分解仍面临巨大挑战。本研究设计并构建了一种双锁结构(NiFeP@γ-NiFeOOH(PO)),通过以下创新策略解决了γ-NiFeOOH催化剂稳定性差的问题:层间锁定机制:利用NiFeP核在电化学活化过程中原位释放形成的磷酸根离子(PO43-),在γ-NiFeOOH层间形成刚性四面体支柱结构,有效阻止层间坍塌并通过静电作用咬合相邻Ni(Fe)O2层板。界面锁定机制:重构的活性层与导电基底之间形成共价键锚定,防止活性物质脱落。打破Bode循环:双锁策略从根本上抑制了γ相向β/α相的结构退化,使γ-NiFeOOH在工业级操作条件下保持结构完整性。该催化剂在1 M KOH中仅需236 mV过电位即可达到100 mA cm-2的电流密度,并在900 mA cm-2下稳定运行超过2500小时,性能远超同类NiFe基催化剂。Fig. 1. The proposed dual-locking strategy versus the conventional evolution pathway. (a) Schematic showing the structural instability and regression in the conventional evolution pathway of γ-MOOH (M=Ni, Fe) derived from M-LDH. (b) Illustration of the proposed dual-locking architecture, where the γ-phase is stabilized via tetrahedral PO43- motifs (interlocking) and interfacial stabilization via strong terminal M-O bonds anchored to the substrate. Copyright 2026, Elsevier Inc.
NiFe基层状双氢氧化物(M-LDH)是目前碱性OER最具前景的非贵金属催化剂之一。在电化学活化过程中,NiFe-LDH会经历结构重构,形成不同的(氧)氢氧化物相。其中,γ-NiFeOOH因其扩展的层间距和高度氧化的金属中心,被认为是OER最高内在活性的催化相。然而,γ-NiFeOOH的形成和稳定面临两大根本挑战:1,动力学限制:从NiFe-LDH前驱体转变为长程有序γ相结构需要高能晶格重构,涉及耦合的质子-电子转移、广泛脱质子化和金属-氧八面体重排。2,热力学不稳定性:经典的Bode循环理论表明,γ相在高氧化态和较大层间距下容易向能量更低的β/α相回归。Fig. 1 展示了文章的核心设计逻辑。常规M-LDH在阳极偏压下可经历β-MOOH到γ-MOOH的结构演化,但由于平面型阴离子和水分子难以稳定支撑扩大的层间结构,γ相容易发生回退和坍塌。传统方法(如成分调控、缺陷工程等)主要修饰局部电子环境,无法从根本上解决层间不稳定问题。本文创新性地提出双重锁定策略,在两个层次同时施加结构约束:层间由四面体PO43-插层支撑,界面由终端M-O键锚定在NiFeP基底上。这样的设计相当于给γ-NiFeOOH同时加上“层间铆钉”和“底部固定桩”,从源头上抑制Bode循环的可逆回退,为稳定亚稳态高活性催化相提供了新思路。双重锁定策略:从“容易回退”的γ相到“被锁住”的γ相
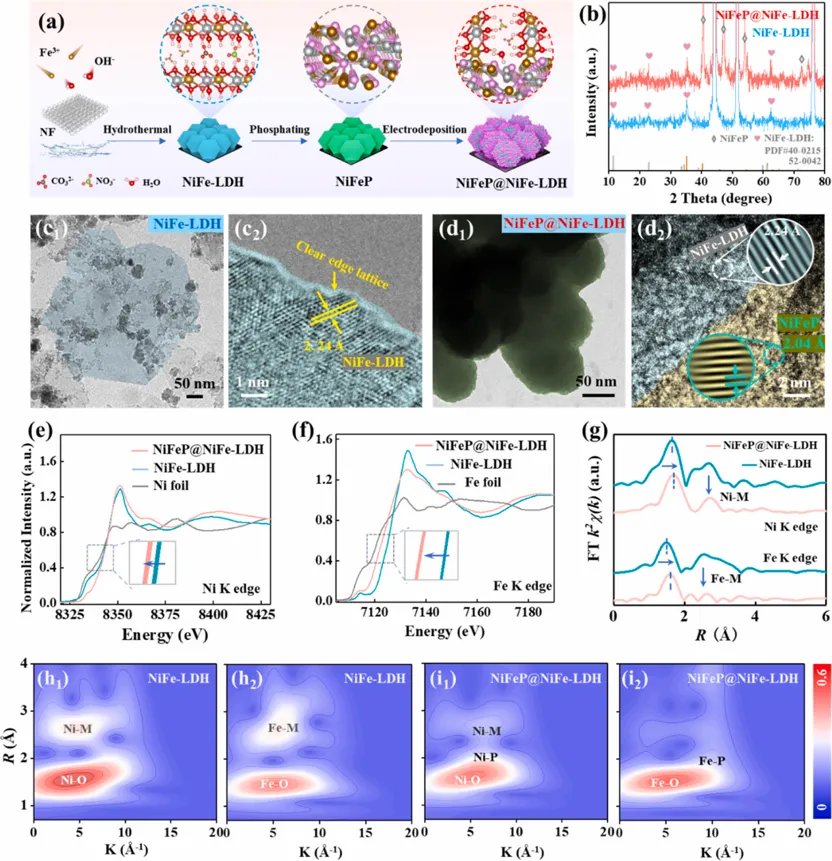
合成方法与前驱体结构
Fig. 2.Synthesis and structural characterization of the NiFeP@NiFe-LDH precursor. (a) Schematic illustration of the synthesis process. (b) XRD patterns of pristine NiFe-LDH and NiFeP@NiFe-LDH. (c, d) TEM and HRTEM images of (c1, c2) NiFe-LDH and (d1, d2) NiFeP@NiFe-LDH. Insets: the corresponding inverse fast Fourier transform (IFFT) images. (e, f) Ni K-edge (e) and Fe K-edge (f) XANES spectra of NiFe-LDH, NiFeP@NiFe-LDH, and reference samples. (g) FT-EXAFS spectra at the Ni and Fe K-edges for NiFe-LDH and NiFeP@NiFe-LDH. (h, i) Wavelet transform (WT)-EXAFS contour maps of the Ni K-edge (h1, i1) and Fe K-edge (h2, i2) for NiFe-LDH and NiFeP@NiFe-LDH, respectively. Copyright 2026, Elsevier Inc.
文章采用三步法构筑NiFeP@NiFe-LDH核壳前驱体:首先,以泡沫镍为基底和Ni源,通过水热反应生长垂直排列的NiFe-LDH纳米片;随后,经NaH2PO2低温磷化,将NiFe-LDH转化为多孔NiFeP纳米片,构建导电的“磷库”内核;最后,通过电沉积在NiFeP表面重新生长NiFe-LDH外壳,得到NiFeP@NiFe-LDH异质结构。结构表征证明,该材料同时具备两个关键前提:其一,NiFeP内核具有较好的导电性,并可在后续阳极活化中释放P物种;其二,外层NiFe-LDH可作为可重构活性层,在OER过程中转化为NiFeOOH。XAS结果显示,NiFeP@NiFe-LDH中Ni和Fe的吸收边相较于NiFe-LDH负移,说明磷化物内核向外层注入电子,形成较为“松弛”的金属-氧配位环境;WT-EXAFS中出现Ni/Fe-P相互作用,也为后续M-P键氧化断裂、P物种迁移和磷酸根支柱形成埋下伏笔。OER性能:低过电位与安培级长寿命并存
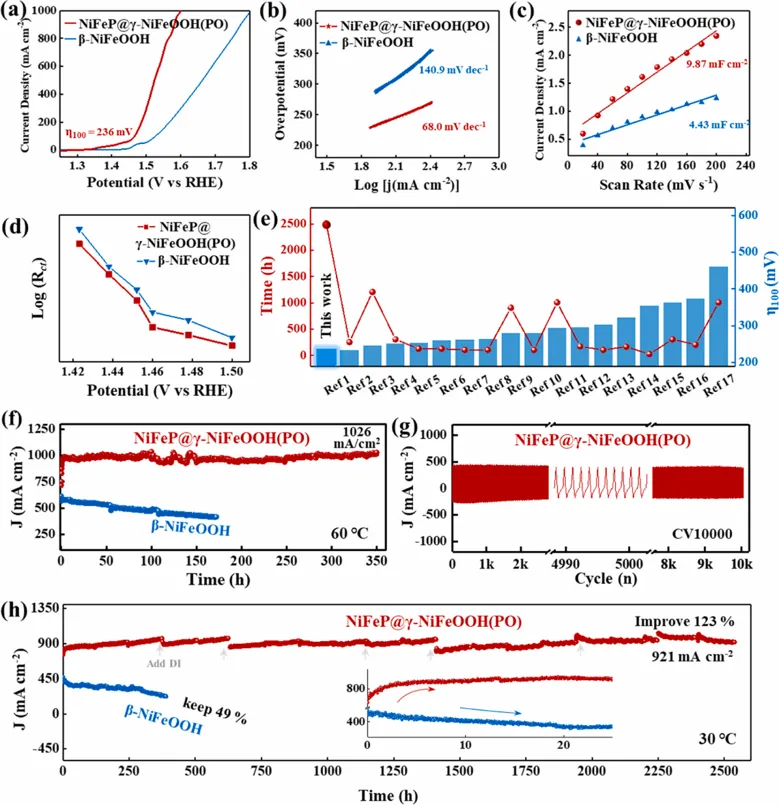
Fig. 3.OER performance of the activated catalysts. (a) Polarization curves of β-NiFeOOH and NiFeP@γ-NiFeOOH(PO). (b) The corresponding Tafel slopes. (c) Double-layer capacitance (Cdl) representing the electrochemically active surface area (ECSA). (d) Voltage-dependent charge transfer resistance (Rct) obtained from EIS fitting. (e) Performance benchmark comparing the overpotential and lifespan of NiFeP@γ-NiFeOOH(PO) against state-of-the-art NiFe-based electrocatalysts. (f) Chronoamperometric stability profiles recorded at 60 ℃, 0.76 V vs. Ag/AgCl without iR compensation. (g) CV test of NiFeP@γ-NiFeOOH(PO) for 10,000 cycles. (h) Chronoamperometric stability profiles recorded at 30℃, 0.8 V vs. Ag/AgCl without iR compensation. The inset highlights the stability retention during the initial 25 h. Copyright 2026, Elsevier Inc.电化学测试显示,NiFeP@γ-NiFeOOH(PO)在1 M KOH中表现出显著优于对照样的OER活性。相较于由NiFe-LDH重构得到的β-NiFeOOH,NiFeP@γ-NiFeOOH(PO)在100、500和1000 mA cm-2时的过电位分别低至236、293和370 mV,明显优于NiFe-LDH、NiFeP、NiFeP@NiFe-LDH前驱体和商业IrO2。其Tafel斜率为68 mV dec-1,说明反应动力学得到明显加快。更值得注意的是稳定性。NiFeP@γ-NiFeOOH(PO)可在约900 mA cm-2 (30 °C) 条件下稳定运行超过2500 h;在约1000 mA cm-2 (60 °C)条件下稳定运行超过350 h;在约1500 mA cm-2 (30 °C)条件下稳定运行超过250 h;同时,在经历10000圈CV和210次循环启停测试后,活性不衰减。相比之下,β-NiFeOOH在相同条件下电流持续衰减,说明单纯形成NiFeOOH并不足以保证高电流长期稳定,关键在于能否稳定高活性γ相。电化学重构:为什么NiFeP@NiFe-LDH会走向γ相?
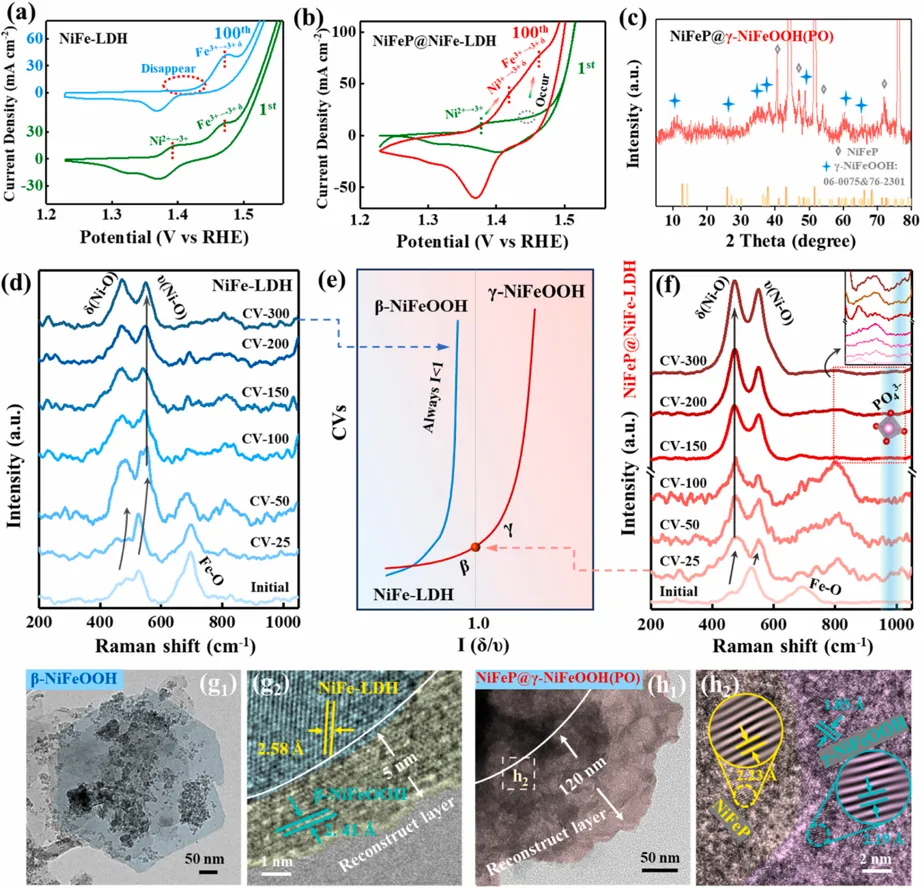
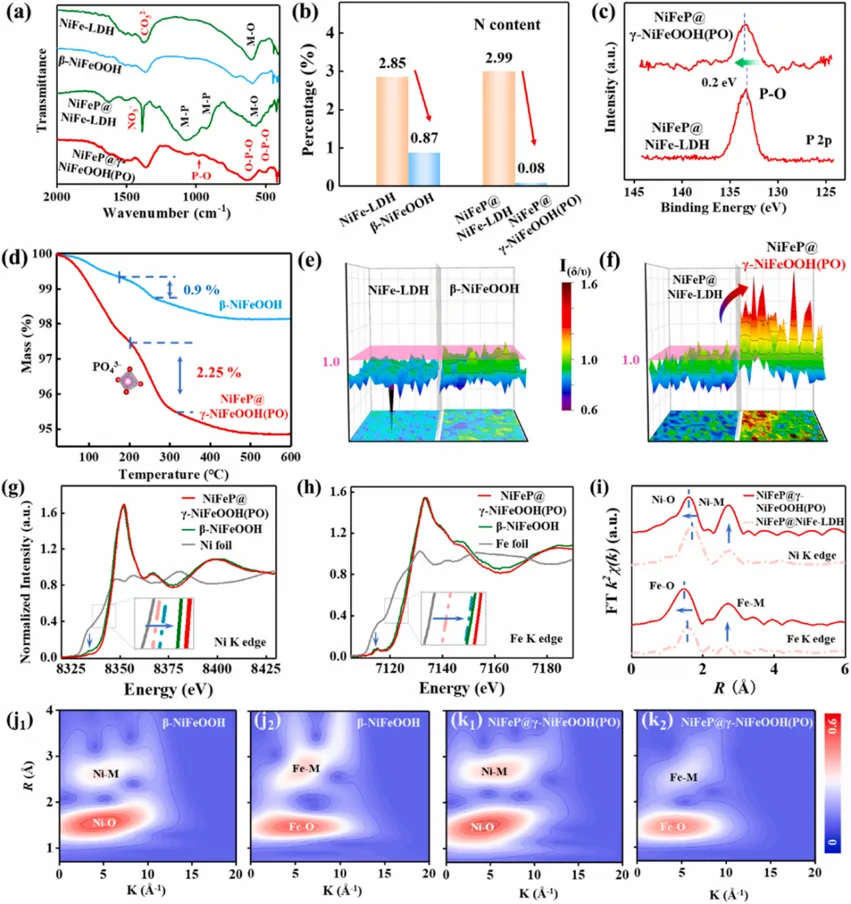
Fig. 4. Phase evolution during electrochemical activation. (a, b) The CV curves of (a) NiFe-LDH and (b) NiFeP@NiFe-LDH recorded at the 1st and 100th cycles. (c) XRD pattern of NiFeP@NiFe-LDH collected after 100 CV cycles (denoted as NiFeP@γ-NiFeOOH(PO)). (d, f) In situ Raman spectra evolution of (d) NiFe-LDH and (f) NiFeP@NiFe-LDH recorded at various cycle intervals. Inset in (f): Magnified view of the region highlighted by the red box. (e) Evolution of the Raman intensity ratio (Iδ/ν) as a function of cycle number, representing the kinetics of the phase transition. Iδ/ν corresponds to the ratio of the δ(Ni-O)-to-ν(Ni-O). (g, h) Post-activation structural characterization: (g1) TEM and (g2) HRTEM images of the activated NiFe-LDH (identified as β-NiFeOOH); (h1) TEM and (h2) HRTEM images of the activated NiFeP@γ-NiFeOOH(PO). Insets: the corresponding IFFT images. Copyright 2026, Elsevier Inc.原位/准原位结构表征揭示了两个前驱体截然不同的重构路径。普通NiFe-LDH在活化后主要形成β-NiFeOOH,重构只局限于边缘浅层区域;而NiFeP@NiFe-LDH在活化过程中出现明显的γ-NiFeOOH特征峰。XRD显示,活化后的NiFeP@γ-NiFeOOH(PO)出现10.6°, 37.5°, 48.4°和66.2°等可归属于γ-NiFeOOH的衍射峰,而NiFe-LDH对照样未出现明显γ相信号。Raman结果提供了更加直接的相判据:Ni-O弯曲振动与伸缩振动强度比Iδ/ν大于1时,通常对应层间距较大且无序度较高的γ-NiOOH结构。NiFeP@NiFe-LDH在约25圈CV活化后Iδ/ν超过1,而NiFe-LDH始终变化很小。此外,NiFeP支持体系中出现约985 cm-1的PO43-振动峰,说明NiFeP基底释放出的P物种进入重构层间,并与γ相形成过程同步发生。TEM和EDS进一步证明,NiFeP@γ-NiFeOOH(PO)形成约120 nm厚的深度重构层,且P元素在重构区域中均匀分布。组成演化与锁相机制:PO43-既是“支柱”也是“电子调节器”
Fig. 5. Phase-locking mechanism and electronic structure modulation. (a) FT-IR spectra of the indicated samples. (b) Comparison of the nitrogen content. (c) High-resolution P 2p XPS spectra. (d) Thermogravimetric analysis (TG) curves. (e, f) Raman mapping images visualizing the Iδ/ν intensity ratio distribution for (e) the NiFe-LDH to β-NiFeOOH transition and (f) the NiFeP@NiFe-LDH to NiFeP@γ-NiFeOOH(PO) transition. (g, h) Normalized Ni K-edge (g) and Fe K-edge (h) XANES spectra of β-NiFeOOH and NiFeP@γ-NiFeOOH(PO). Inset: Magnified view of the absorption edge region. (Note: Dashed lines representing the precursors from Fig. 2 are included for reference). (i) FT-EXAFS spectra at the Ni and Fe K-edges for the relevant samples. (j, k) Wavelet transform (WT)-EXAFS contour maps of the Ni K-edge (j1, k1) and Fe K-edge (j2, k2) for (j) the reference NiFe-LDH and (k) the NiFeP@γ-NiFeOOH(PO). Copyright 2026, Elsevier Inc.FT-IR、XPS、XAS和TG分析共同证明了PO43-插层的存在及其稳定作用。活化前,NiFe-LDH和NiFeP@NiFe-LDH均含有CO32-、NO3-等层间平面型阴离子;活化后,NiFe-LDH中这些阴离子信号明显减弱,表明其逐渐流失,而NiFeP@γ-NiFeOOH(PO)则出现P-O (1064 cm-1)和O-P-O (620、510 cm-1)等振动信号,说明四面体磷酸根进入层间。P 2p XPS正移约0.2 eV,也支持P物种转化为氧化态磷酸根。更关键的是,PO43-不仅稳定结构,还调节电子态。XANES表明,NiFeP@γ-NiFeOOH(PO)在活化后Ni和Fe吸收边显著正移,代表金属中心价态升高;XPS中Ni 2p和Fe 2p结合能分别正移0.54和0.70 eV,O 1s负移0.21 eV,说明金属中心电子被抽离,金属-氧共价性增强,有利于提高OER性能。换句话说,磷酸根不是一个“惰性插层客体”,而是同时承担结构加固与电子调控的双重角色。Fig. 6. Proposed atomistic mechanisms for the electrochemical phase reconstruction. (a) The conventional reconstruction pathway involving the transition from NiFe-LDH to β-NiFeOOH. (b) The proposed dual-locking reconstruction pathway, showing the transition from the NiFeP@β-NiFeOOH to NiFeP@γ-NiFeOOH (PO) via phosphate intercalation and interface anchoring. Copyright 2026, Elsevier Inc.作者进一步提出了统一的原子尺度重构机制。对于普通NiFe-LDH,在阳极偏压下,端基-OH脱质子化,层间CO32-和NO3-等平面型阴离子逐渐脱出,层间失去支撑,最终形成层间距小,低价的β-NiFeOOH。由于缺乏额外支撑,这一路径更接近Bode循环中热力学稳定相的回落。对于NiFeP@NiFe-LDH,初始脱质子化过程类似,但NiFeP内核在高电位下发生M-P键氧化断键,释放P物种。这些P物种迁移到NiFeOOH层间并转化为高电荷密度的四面体PO43-,其刚性构型与相邻Ni(Fe)O2层板形成类似“锯齿状咬合”的层间互锁,此外,高电荷密度的PO43-插入层间,可以提高Ni/Fe的价态(通过静电补偿);同时重构层通过界面M-O键锚定在NiFeP基底上,形成基底锁定。层间互锁阻止结构坍塌,基底锁定抑制活性层剥离,两者共同打断Bode循环的可逆性,使亚稳γ相能够在高电流和长时间运行中保持。DFT计算:高活性与高稳定性的微观来源
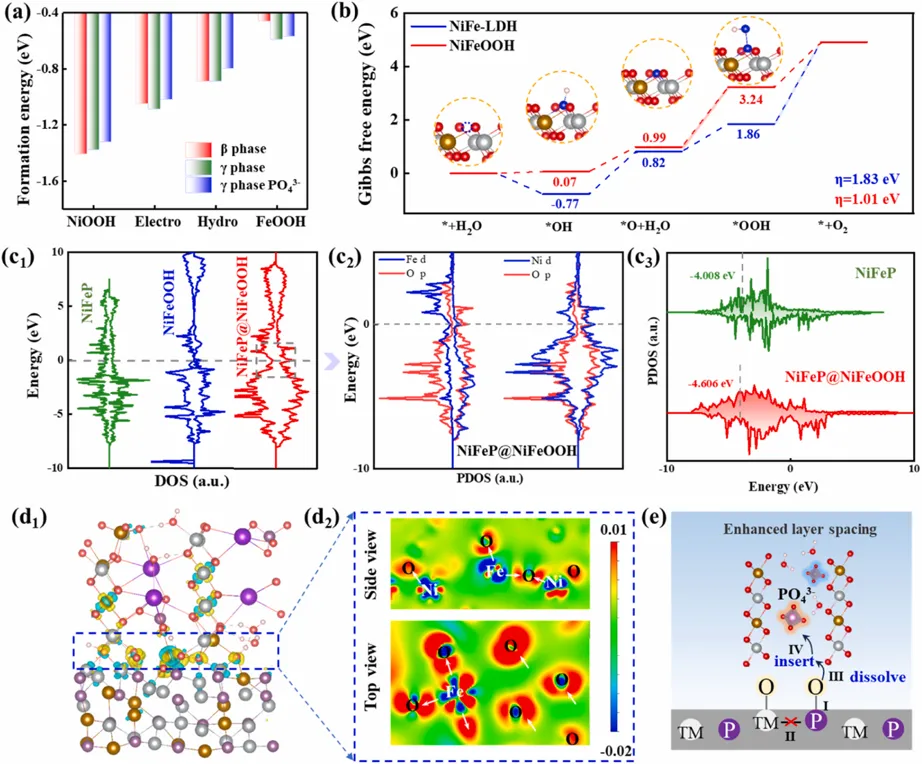 Fig. 8. DFT insights into the activity origin and phase stability. (a) Calculated formation energies of various Ni1-xFexOOH models. (Note: "Electro" and "Hydro" refer to models constructed based on electrodeposition and hydrothermal experimental data, respectively). (b) Gibbs free energy diagrams for the OER intermediates (*OH, *O, and *OOH) on the surfaces of NiFe-LDH and the activated heterostructure. (c1) Total density of states (TDOS) for NiFeP, NiFeOOH, and the NiFeP@NiFeOOH interface. (c2) Projected density of states (PDOS) showing the orbital overlap between Fe/Ni 3d and O 2p states near the Fermi level. (c3) Comparison of the d-band center positions in NiFeP and NiFeP@NiFeOOH. (d1) Charge density difference plot at the NiFeP@NiFeOOH interface. Yellow and green isosurfaces indicate electron accumulation and depletion, respectively. (d2) 2D cross-sectional charge density distribution corresponding to the area in the dotted frame. (e) Schematic illustration of the proposed structural evolution and stabilization mechanism. Copyright 2026, Elsevier Inc.DFT计算从反应能垒和电子结构两个角度解释了性能提升。对于β-NiFeOOH,OER过程中关键步骤的能垒较高,达到1.81 eV;而在γ-NiFeOOH(PO)中,决速步骤转移为*O到*OOH的转化,能垒降低至1.01 eV,说明PO43-插层能够显著优化含氧中间体吸附和反应路径。态密度分析表明,NiFeP@γ-NiFeOOH(PO)在费米能级附近具有更高的电子态密度,有利于界面电荷传输;Ni/Fe 3d与O 2p轨道杂化增强,表明M-O共价性提高。d带中心由NiFeP的-4.008 eV下移至NiFeP@γ-NiFeOOH(PO)的-4.606 eV,从而缓和关键中间体过强吸附,降低整体反应能垒。电荷密度差分进一步显示,Ni/Fe中心与磷酸根配位氧之间存在明显电荷重分布,说明PO43-支柱可通过共价和静电作用共同稳定扩大的γ相层间结构。这项工作围绕NiFe基OER催化剂中“γ相活性高但难稳定”的核心矛盾,提出了兼具结构约束和电子调控功能的双重锁定策略。通过NiFeP@NiFe-LDH前驱体的可控电化学重构,作者实现了自源性PO43-插层和基底界面锚定的协同构筑,使亚稳态γ-NiFeOOH在安培级水氧化条件下长期保持。与传统的元素掺杂、缺陷调控或单纯提高导电性不同,该策略直接瞄准γ-NiFeOOH不稳定的根源,即层间坍塌和Bode循环退行。PO43-四面体像支柱一样互锁层间,同时诱导Ni/Fe中心更高价态和更强M-O共价性;NiFeP基底则提供导电通道、P源和界面锚定。最终得到的NiFeP@γ-NiFeOOH(PO)在100 mA cm-2下仅需236 mV过电位,并可在约900 mA cm-2 (30 °C) 条件下稳定运行超过2500 h;在约1000 mA cm-2 (60 °C)条件下稳定运行超过350 h;在约1500 mA cm-2 (30 °C)条件下稳定运行超过250 h;同时,在经历10000圈CV和210次循环启停测试后,活性不衰减。该研究不仅为NiFe基OER催化剂的工业级长寿命设计提供了新思路,也为其它亚稳高活性催化相的稳定化提供了可借鉴的结构工程原则。黄维清 教授:博士、教授、博士生导师。主持或主研国家自然科学基金项目、湖南省自然科学基金项目、湖南省科技计划项目等课题十余项。已在Adv. Mater., Nano Lett.,ACS Nano,Adv. Funct. Mater.,Appl. Catal. B: Environ.,Phys. Rev. B,APL等杂志发表论文200余篇,SCI引用9900余次,H 因子53。主要研究方向为计算凝聚态物理、清洁能源材料物理及器件、微纳结构光电功能材料物理及清洁能源和绿色环境材料。黄桂芳 教授:博士、教授、博士生导师。主持或主研国家自然科学基金项目、湖南省自然科学基金项目、湖南省科技计划项目等课题十余项。已在Adv. Mater.、ACS Nano、Adv. Funct. Mater.、Appl. Catal. B: Environ.和Electrochim Acta 等学术杂志上发表论文100多篇;获批国际专利一项,发明专利两项,实用新型专利两项,起草国家标准两项、机械行业标准两项。万惠 讲师:博士、硕士生导师。主持长沙市自然科学基金项目、湖南省自然科学基金项目和湖南省教育厅科研项目各1项,参与国家自然科学基金项目3项。迄今已在Adv. Mater.,Nano Lett.,Adv. Energy Mater.,Adv. Func. Mater., Appl. Catal. B: Environ.和Nano Energy等期刊发表论文38篇。长期从事清洁能源纳米材料的设计、制备与理论计算,在功能材料性能模拟、界面特性调控与可控制备等方面形成了系统的研究体系,兼具扎实的理论与实验基础。M.-H. Xian, M.-Y. Xie, Y. Luo, J. Nie, J.-R. Huang, G.-F. Huang, H. Wan, W. Hu, W.-Q. Huang, Breaking the Bode cycle: Stabilizing metastable γ-NiFeOOH via a dual-locking strategy for ampere-level water oxidation, Appl Catal, B Environ., 400 (2026) 127185
Fig. 8. DFT insights into the activity origin and phase stability. (a) Calculated formation energies of various Ni1-xFexOOH models. (Note: "Electro" and "Hydro" refer to models constructed based on electrodeposition and hydrothermal experimental data, respectively). (b) Gibbs free energy diagrams for the OER intermediates (*OH, *O, and *OOH) on the surfaces of NiFe-LDH and the activated heterostructure. (c1) Total density of states (TDOS) for NiFeP, NiFeOOH, and the NiFeP@NiFeOOH interface. (c2) Projected density of states (PDOS) showing the orbital overlap between Fe/Ni 3d and O 2p states near the Fermi level. (c3) Comparison of the d-band center positions in NiFeP and NiFeP@NiFeOOH. (d1) Charge density difference plot at the NiFeP@NiFeOOH interface. Yellow and green isosurfaces indicate electron accumulation and depletion, respectively. (d2) 2D cross-sectional charge density distribution corresponding to the area in the dotted frame. (e) Schematic illustration of the proposed structural evolution and stabilization mechanism. Copyright 2026, Elsevier Inc.DFT计算从反应能垒和电子结构两个角度解释了性能提升。对于β-NiFeOOH,OER过程中关键步骤的能垒较高,达到1.81 eV;而在γ-NiFeOOH(PO)中,决速步骤转移为*O到*OOH的转化,能垒降低至1.01 eV,说明PO43-插层能够显著优化含氧中间体吸附和反应路径。态密度分析表明,NiFeP@γ-NiFeOOH(PO)在费米能级附近具有更高的电子态密度,有利于界面电荷传输;Ni/Fe 3d与O 2p轨道杂化增强,表明M-O共价性提高。d带中心由NiFeP的-4.008 eV下移至NiFeP@γ-NiFeOOH(PO)的-4.606 eV,从而缓和关键中间体过强吸附,降低整体反应能垒。电荷密度差分进一步显示,Ni/Fe中心与磷酸根配位氧之间存在明显电荷重分布,说明PO43-支柱可通过共价和静电作用共同稳定扩大的γ相层间结构。这项工作围绕NiFe基OER催化剂中“γ相活性高但难稳定”的核心矛盾,提出了兼具结构约束和电子调控功能的双重锁定策略。通过NiFeP@NiFe-LDH前驱体的可控电化学重构,作者实现了自源性PO43-插层和基底界面锚定的协同构筑,使亚稳态γ-NiFeOOH在安培级水氧化条件下长期保持。与传统的元素掺杂、缺陷调控或单纯提高导电性不同,该策略直接瞄准γ-NiFeOOH不稳定的根源,即层间坍塌和Bode循环退行。PO43-四面体像支柱一样互锁层间,同时诱导Ni/Fe中心更高价态和更强M-O共价性;NiFeP基底则提供导电通道、P源和界面锚定。最终得到的NiFeP@γ-NiFeOOH(PO)在100 mA cm-2下仅需236 mV过电位,并可在约900 mA cm-2 (30 °C) 条件下稳定运行超过2500 h;在约1000 mA cm-2 (60 °C)条件下稳定运行超过350 h;在约1500 mA cm-2 (30 °C)条件下稳定运行超过250 h;同时,在经历10000圈CV和210次循环启停测试后,活性不衰减。该研究不仅为NiFe基OER催化剂的工业级长寿命设计提供了新思路,也为其它亚稳高活性催化相的稳定化提供了可借鉴的结构工程原则。黄维清 教授:博士、教授、博士生导师。主持或主研国家自然科学基金项目、湖南省自然科学基金项目、湖南省科技计划项目等课题十余项。已在Adv. Mater., Nano Lett.,ACS Nano,Adv. Funct. Mater.,Appl. Catal. B: Environ.,Phys. Rev. B,APL等杂志发表论文200余篇,SCI引用9900余次,H 因子53。主要研究方向为计算凝聚态物理、清洁能源材料物理及器件、微纳结构光电功能材料物理及清洁能源和绿色环境材料。黄桂芳 教授:博士、教授、博士生导师。主持或主研国家自然科学基金项目、湖南省自然科学基金项目、湖南省科技计划项目等课题十余项。已在Adv. Mater.、ACS Nano、Adv. Funct. Mater.、Appl. Catal. B: Environ.和Electrochim Acta 等学术杂志上发表论文100多篇;获批国际专利一项,发明专利两项,实用新型专利两项,起草国家标准两项、机械行业标准两项。万惠 讲师:博士、硕士生导师。主持长沙市自然科学基金项目、湖南省自然科学基金项目和湖南省教育厅科研项目各1项,参与国家自然科学基金项目3项。迄今已在Adv. Mater.,Nano Lett.,Adv. Energy Mater.,Adv. Func. Mater., Appl. Catal. B: Environ.和Nano Energy等期刊发表论文38篇。长期从事清洁能源纳米材料的设计、制备与理论计算,在功能材料性能模拟、界面特性调控与可控制备等方面形成了系统的研究体系,兼具扎实的理论与实验基础。M.-H. Xian, M.-Y. Xie, Y. Luo, J. Nie, J.-R. Huang, G.-F. Huang, H. Wan, W. Hu, W.-Q. Huang, Breaking the Bode cycle: Stabilizing metastable γ-NiFeOOH via a dual-locking strategy for ampere-level water oxidation, Appl Catal, B Environ., 400 (2026) 127185https://doi.org/10.1016/j.apcatb.2026.127185